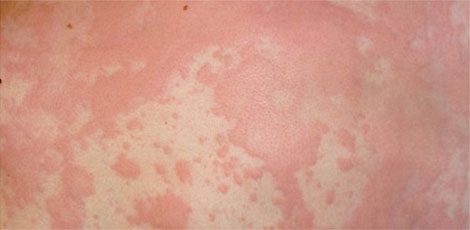
Crises de inchaço podem acontecer de forma espontânea ou decorrentes de traumas
Foto: internet
Mariana DantasDo NE10
A estudante Larissa Peres Paulino, de 17 anos, tinha apenas dez meses de vida quando teve a sua primeira crise. O seu bracinho ficou completamente inchado e a mãe a levou para o hospital. O diagnóstico dado na época foi uma crise alérgica. Os anos se passaram e Larissa continuou sofrendo com os edemas, que apareciam na face, mãos, pés e na região do abdômen. Foram várias as doses de adrenalina que recebeu nas emergências das unidades de saúde para amenizar a “suposta reação alérgica”. Na adolescência os sintomas aumentaram e um simples botão de uma calça jeans apertada provocava um edema em sua barriga. O diagnóstico correto só foi descoberto no início deste ano. Larissa possui angioedema hereditário (AEH), doença grave considerada rara, que ainda não tem cura, cuja incidência é de um caso para cada 50 mil habitantes no mundo.
O angioedema hereditário (angio = vaso sanguíneo e edema = inchaço) é uma doença genética, que atinge homens e mulheres e geralmente apresenta os primeiros sintomas ainda na infância. Nos portadores de AEH, há uma alteração no gene que produz a proteína chamada inibidor de C1-esterase, que controla os sistemas complemento, de contato e das cininas. Esses sistemas atuam promovendo o processo inflamatório no organismo. Com a deficiência do inibidor de C1, a inflamação, que pode ser desencadeada até por pequenos traumas, se apresenta de forma descontrolada. Trata-se de uma disfunção no sistema imunológico provocada pela falta do inibidor de uma das proteínas do sangue responsável por combater inflamações e infecções. Esse inibidor é chamado de C1 esterase. Se uma pessoa sem a doença levar uma pancada, normalmente surgirá apenas um inchaço no local. Já no portador de AEH, esse edema crescerá bastante.
Por se tratar de uma doença de herança genética, filhos de pacientes afetados têm 50% de chance de herdar a patologia. No entanto, em 20% dos casos pode ocorrer mutações genéticas recentes, não sendo identificado outros familiares afetados.
O angioedema hereditário (angio = vaso sanguíneo e edema = inchaço) é uma doença genética, que atinge homens e mulheres e geralmente apresenta os primeiros sintomas ainda na infância. Nos portadores de AEH, há uma alteração no gene que produz a proteína chamada inibidor de C1-esterase, que controla os sistemas complemento, de contato e das cininas. Esses sistemas atuam promovendo o processo inflamatório no organismo. Com a deficiência do inibidor de C1, a inflamação, que pode ser desencadeada até por pequenos traumas, se apresenta de forma descontrolada. Trata-se de uma disfunção no sistema imunológico provocada pela falta do inibidor de uma das proteínas do sangue responsável por combater inflamações e infecções. Esse inibidor é chamado de C1 esterase. Se uma pessoa sem a doença levar uma pancada, normalmente surgirá apenas um inchaço no local. Já no portador de AEH, esse edema crescerá bastante.
Por se tratar de uma doença de herança genética, filhos de pacientes afetados têm 50% de chance de herdar a patologia. No entanto, em 20% dos casos pode ocorrer mutações genéticas recentes, não sendo identificado outros familiares afetados.

As crises de inchaço podem acontecer de forma espontânea ou decorrentes de fatores desencadeantes como trauma (pancada), infecção, cirurgia e até alterações hormonais ou estresse. “Os edemas ocorrem de forma recorrente atingindo principalmente as pálpebras, lábios e língua, mãos, pés e órgãos genitais. Também pode ocorrer na região abdominal, além de edema na laringe, que pode levar ao fechamento das vias áreas, conhecido como edema de glote, ocasionando morte por asfixia em cerca de 30% dos casos não tratados”, explica a médica Almerinda Maria Rego Silva, responsável pelo ambulatório de imunologia clinica do Hospital das Clínicas - UFPE, considerado um centro de referência para tratamento da doença em Pernambuco.
O inchaço que atinge a face, os genitais, as mãos e os pés é geralmente doloroso sem coceira, desfigurante e debilitante para os pacientes, com duração media de 2 a 5 dias. No abdômen, a dor intensa pode estar associada a náusea, vômito e diarreia e, muitas vezes, é confundida com quadros agudos e os pacientes são submetidos a cirurgias desnecessárias.
O inchaço que atinge a face, os genitais, as mãos e os pés é geralmente doloroso sem coceira, desfigurante e debilitante para os pacientes, com duração media de 2 a 5 dias. No abdômen, a dor intensa pode estar associada a náusea, vômito e diarreia e, muitas vezes, é confundida com quadros agudos e os pacientes são submetidos a cirurgias desnecessárias.

Atualmente, cerca de 20 pessoas estão em tratamento no HC/UFPE. “O tratamento de prevenção às crises é realizado por via oral. A medicação deve ser tomada diariamente e é oferecida pelo Sistema Único de Saúde”, explica a médica Almerinda. Para combater as crises agudas, é preciso uma medicação injetável que é autorizada no Brasil desde 2011, mas ainda não é fornecida pelo SUS. Essa medicação, de custo elevado, contém a substancia ativa icatibanto,que atua bloqueando os receptores da bradicinina. Cada injeção custa, em média, R$ 3 mil e, dependendo da crise, o paciente pode receber até três doses do medicamento. Para conseguir a injeção, os pacientes de todo o Brasil precisam recorrer à Justiça.

“É um desgaste para os portadores de AEH. Estamos lutando juntamente com os médicos para que a medicação seja oferecida pelo SUS. Por enquanto, nos resta orientar as pessoas que nos procuram”, afirma a presidente da Associação Brasileira de Portadores de Angioedema Hereditário (Abranghe), Raquel de Oliveira.
Portadora da doença, Raquel teve a iniciativa de criar na rede social Orkut, no ano de 2005, um grupo para compartilhar informações sobre o AEH. “Como a doença é pouco divulgada, o grupo acabou se tornando uma fonte de informação e serviço para as pessoas. Em 2010, decidimos fundar oficialmente Abranghe (com sede em Campinas – SP)”, explica a presidente da entidade, a única que reúne dados de pacientes no País.
De acordo com a Abranghe, o Brasil possui 691 casos de AEH diagnosticados, sendo 56 na região Nordeste, 447 no Sudeste, 107 no Sul, 39 no Centro Oeste e 12 no Norte. Nos últimos três anos, nove brasileiros já diagnosticados morreram em conseqüência da doença, todos vítimas de edema de glote.
Talvez por ser uma doença rara, o angioedema hereditário é pouco conhecido. Seu diagnóstico pode ser confundido com crises alérgicas. “Pela falta de conhecimento de muitos médicos, sofri muito para descobrir o que realmente tinha”, diz Larissa. Para não correr mais o risco de receber um atendimento inadequado, ela não sai de casa sem a sua carteira de identificação fornecida pela Abranghe e um laudo explicativo da doença. Se precisar receber a injeção, Larissa também leva o medicamento (que recebeu com autorização Judicial) para ser aplicado no hospital.
Os mesmos cuidados também são tomados pelo aposentado Iran Viana de Souza, 58 anos. Diferentemente da maioria dos portadores de AEH, ele apresentou os primeiros sintomas após os 50 anos. O seu caso vem sendo estudado pela equipe do Hospital das Clínicas e pode estar entre os ainda mais raros: quando a origem não é hereditária, mas provocada por uma mutação genética recente. “Os meus dois filhos já fizeram exames de sangue para identificar a carência do inibidor de C1 e, graças a Deus, não possuem a doença. Também não conheço nenhum outro caso na família”, conta Iran Viana.
Depois de ter várias crises, a maioria na região do abdômen, Iran descobriu que era portador da doença no início do ano de 2010. “A minha sorte foi ter descoberto no início daquele ano. Pois em dezembro de 2010, quando ainda não existia a injeção no Brasil, tive um grave edema na língua que quase me levou à morte”, contou Iran. Ele precisou passar por procedimento de traqueostomia e ficou internado durante oito dias na Unidade de Tratamento Intensivo (UTI). “Todo o tratamento foi eficaz porque os médicos já sabiam da minha doença”, concluiu.
Nenhum comentário:
Postar um comentário